:format(webp):quality(90)/https://www.descopera.ro/wp-content/uploads/2020/04/19063542/2-raspandire-coronavirus-cale-evolutie-11-april-2020-profimedia-05.jpg)
:format(jpeg):quality(80)/https://www.descopera.ro/wp-content/uploads/2020/04/19063542/1-harta-raspandire-virus.jpg)
:format(jpeg):quality(80)/https://www.descopera.ro/wp-content/uploads/2020/04/19063542/3-noul-coronaviorus-la-microscop-a-niaid-rml-descopera.jpg)
:format(jpeg):quality(80)/https://www.descopera.ro/wp-content/uploads/2020/04/19063542/4-noul-coronaviorus-la-microscop-niaid-rml-descopera.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1741781917/68436bdca60d682d7f3f2377749b7da9-t.jpg)
Analizând primele 160 de genomuri complete ale virusului SARS-CoV-2, secvenţiate apoi de la pacienţi umani, oamenii de ştiinţă au trasat o parte din răspândirea iniţială a noului coronavirus, prin mutaţiile sale, ceea ce creează filiaţii virale diferite.
„Există prea multe mutaţii rapide pentru a urmări atent un arbore genealogic al COVID-19. Am folosit un algoritm matematic pentru a vizualiza simultan toţi arborii plauzibili”, a afirmat geneticianul Peter Forster, autorul principal al studiului din cadrul Universităţii din Cambridge.
Potrivit doctorului Forster, aceste tehnici sunt folosite în special pentru „cartografierea, prin ADN, a mişcărilor populaţiilor umane preistorice”.
„Credem că este una dintre primele dăţi când s-a folosit pentru urmărirea rutelor de infecţie ale unui coronavirus precum COVID-19”, a precizat acesta.
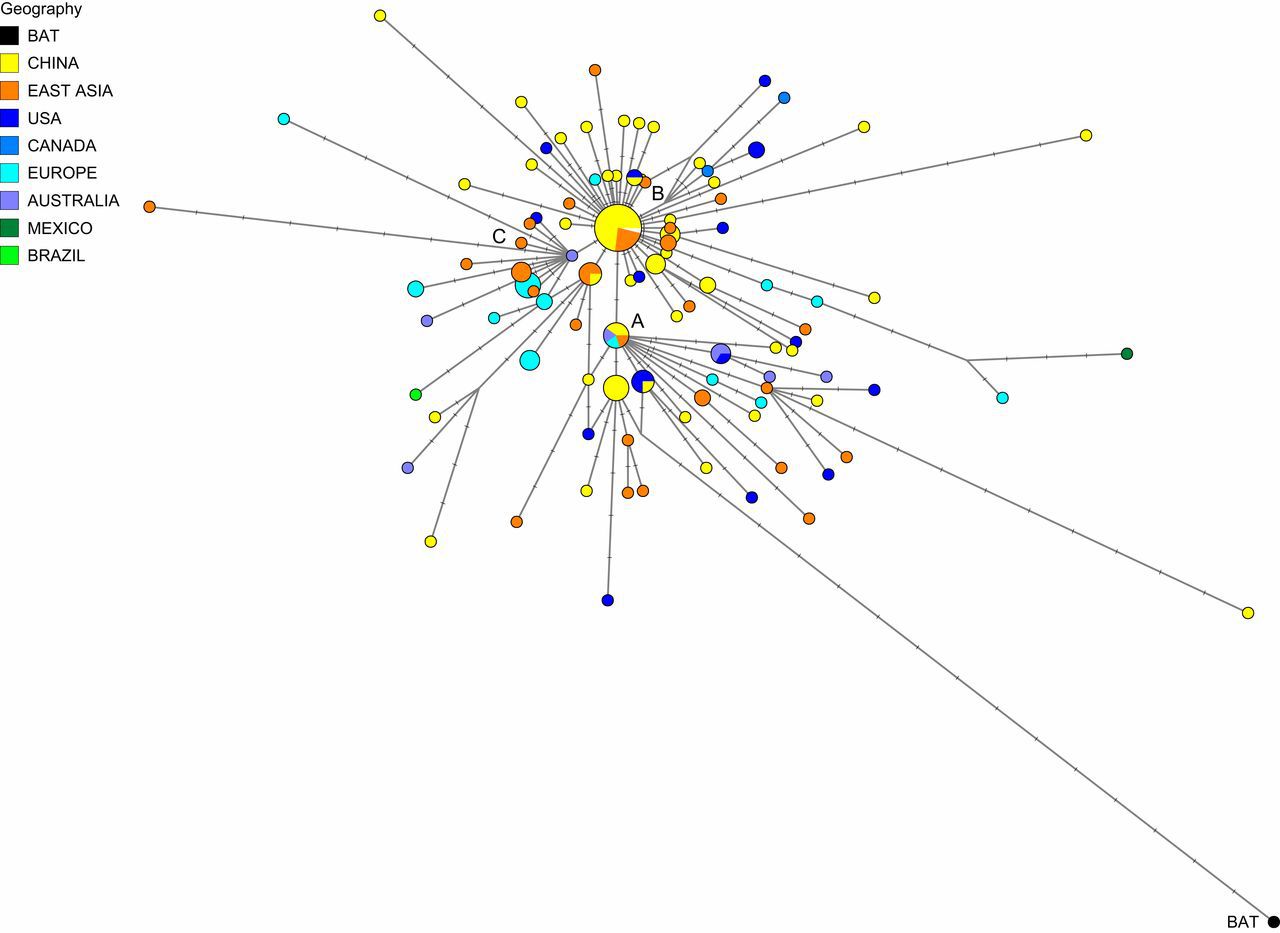
Echipa condusă de geneticianul de la Cambridge a folosit informaţii din mostre de virus prelevate din întreaga lume, între 24 decembrie 2019 şi 4 martie 2020. Cercetarea a evidenţiat „variante” distincte ale COVID-19, constând în grupuri liniare strâns legate între ele, pe care le-au denumit „A”, „B” şi „C”.
Forster şi colegii săi au descoperit că tipul de COVID-19 cel mai apropiat de cel descoperit la lilieci este tipul „A” – „genomul original al virusului uman”- care a fost prezent în Wuhan, dar, spre surprinderea cercetătorilor, nu a fost tipul de virus predominant în oraşul din China.
Au fost observate versiuni mutante ale tipului „A” la americanii despre care s-a raportat că au trăit în Wuhan. Totodată, un număr mare de virusuri de tip A au fost găsite la pacienţi din SUA şi Australia.
Principalul virus din Wuhan a fost tipul „B”, predominant la pacienţii din Asia de Est. Cu toate acestea, varianta B a virusului nu a călătorit mult în afara regiunii, fără mutaţii ulterioare. Potrivit cercetătorilor, a existat „rezistenţă” împotriva acestui tip de coronavirus în afara Asiei de Est.
Varianta „C” a virusului SARS-CoV-2 este principalul tip de coronavirus din Europa, întâlnit la primii pacienţi din Franţa, Italia, Suedia şi Anglia. Această variantă a coronavirusului lipseşte din eşantioanele studiate în China continentală, dar a fost identificată în Singapore, Hong Kong şi Coreea de Sud.
Mai mult, noua analiză sugerează că una dintre primele introduceri ale virusului în Italia a venit prin intermediul primei infecţii din Germania, documentată în 27 ianuarie, iar o altă cale de infecţie timpurie din Italia a fost legată de un „grup de Singapore”.
Credit foto: Peter Forster, Lucy Forster, Colin Renfrew, şi Michael Forster
Mai important, cercetătorii afirmă că tehnicile lor de reţea genetică au urmărit cu exactitate stabilirea căilor de infecţie: mutaţiile şi genealogia virală s-au alăturat în punctele dintre cazurile cunoscute de COVID-19.
Astfel, oamenii de ştiinţă susţin că aceste metode „filogenetice” ar putea fi aplicate la cea mai recentă secvenţiere a genomului coronavirusului, pentru a ajuta la prezicerea viitoarelor puncte fierbinţi de transmitere şi creştere a numărului de cazuri COVID-19, la nivel mondial.
„Analiza reţelei filogenetice are potenţialul de a ajuta la identificarea surselor de infecţie COVID-19 nedocumentate, care pot fi apoi plasate în carantină pentru a controla extinderea suplimentară a bolii în întreaga lume”, a afirmat Forster.
Varianta „A”, cea mai apropiată de virusul detectat nu numai la lilieci, cât şi la pangolini, este descrisă de cercetători ca fiind „rădăcina epidemiei” de coronavirus. Tipul „B” este derivat din „A”, separat de două mutaţii, în timp ce varianta „C” este catalogată de cercetători drept o „fiică” a lui „B”.
Cercetătorii afirmă că localizarea variantei „B” în Asia de Est ar putea fi rezultatul unui „efect fondator”: un blocaj genetic care apare atunci când, în cazul unui virus, un nou tip este stabilit dintr-un grup mic şi izolat de infecţii.
Forster susţine, însă, că există o altă explicaţie care ar putea fi luată în considerare. „Virusul de tipul B din Wuhan ar putea fi adaptat imunologic sau la mediu la o mare parte a populaţiei Asiei de Est. Este posibil să aibă nevoie de mutaţie pentru a depăşi rezistenţa din afara Asiei de Est. Se pare că vedem o rată de mutaţie mai lentă în Asia de Est decât în altă parte, în această fază iniţială”, a afirmat cercetătorul.
Acesta a precizat că reţeaua virală pe care a detaliat-o este imaginea primelor etape ale epidemiei de coronavirus, înainte ca „traseele evolutive ale COVID-19 să devină întunecate de un număr mare de mutaţii”.
Forster spune că cea mai recentă cercetare sugerează că prima infecţie cu noul coronavirus şi răspândirea COVID-19 în rândul oamenilor a avut loc între jumătatea lunii septembrie şi începutul lunii decembrie.
Studiul a fost publicat în jurnalul ştiinţific Proceedings of the National Academy of Sciences (PNAS).
Vă recomandăm să citiţi şi:
Fizicienii de la CERN au făcut un ventilator pe baterii pentru cazurile uşoare de COVID-19
Rafila: Nu orice fel de material este eficient pe post de mască. ”Eşarfele, mai puţin”
:format(jpeg):quality(80)/https://www.descopera.ro/wp-content/uploads/2021/11/mihaela-stoica.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1589795485/e14bc0eea58482b630dd0084c088886b-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1589795423/273998a60dbc578f0ffee9528dfd5f45-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/03/1766046989/cf9d5bb9894deeba45a7f70e2bad1201-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1766046944/da984cfa0bec8ba12515803ff52a04ba-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1687506106/89b94c00c7e2922b71570b99a202802c-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1619101788/a08779d2285a9ccbee06c19e2ed36879-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1604659743/94de1ccc78dd883da232d1adfcd01660-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1741781930/4abf76e59264e6ebae245b7f7456bf0a-t.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/ghosting-psihologie-shutter_Descopera-1024x684.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/senzori-cancer-shutter_Descopera-1024x605.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/alimente-mancare-varsta-shutter_descopera-1024x540.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/ritualuri-noaptea-intuneric-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/epuizat-emotional-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/telefonul-dependenta-shutter_Descopera-1024x683.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1589795438/7160a3930dd543478610eaf5f3699649-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2024/01/1623146229/eee2fda331856b8548658339f0001221-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2023/11/1589795452/715050791c680c518054354f939709bf-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/04/1593777592/5817cab649a4e490c88402d426e601b3-t.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/ochii-vertebratelor_shutterstock_Descopera-10-1024x576.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/reptila-mumificata-wiki_Descopera-1024x559.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/caracatitele-specii-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/ghosting-psihologie-shutter_Descopera-1024x684.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/senzori-cancer-shutter_Descopera-1024x605.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2025/04/cea-mai-veche-tara-din-lume_shutterstock_Descopera-2-1024x739.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/alimente-mancare-varsta-shutter_descopera-1024x540.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2025/12/slabit-obezitate-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/antioxidant-sanatate-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/ziua-din-saptamana-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2025/04/iuri-gagarin-wiki_Descopera-1024x640.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/mesaje-de-paste-shutter_descopera-3-1024x682.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/wi-fi-internet-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/02/bacterii-microbiom-intestin-shutter_descopera-1024x751.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/vremea-prognoza-meteo-shutter_Descopera-1024x588.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/hotel-europa-shutter_Descopera-1024x659.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/04/ritualuri-noaptea-intuneric-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/02/Regele-Carol-si-Regina-Elisabeta-mnir_Descopera-1024x790.jpg)